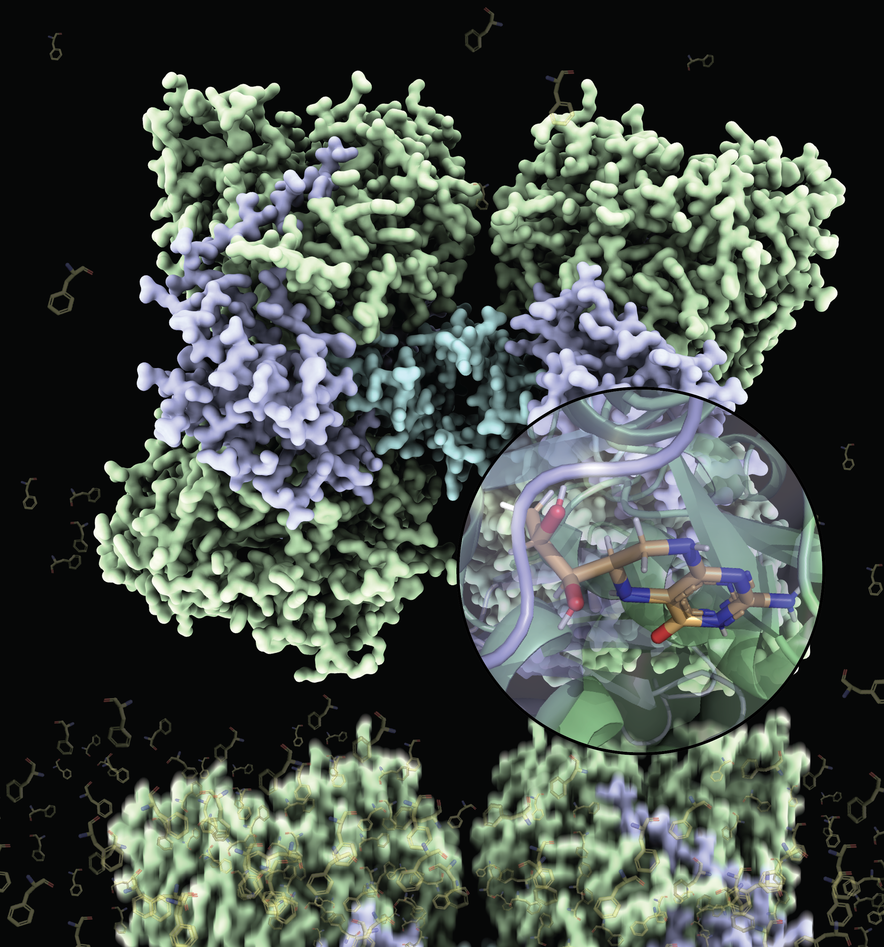
The structure of the enzyme implicated in phenylketonuria (PKU)
The structure of full-length phenylalanine hydroxylase in complex with the cofactor BH4 is presented in the most recent issue of PNAS. As the cofactor is also used as a therapy for PKU, this structure is of medical importance.
Main content
Two decades after the first partial structures of human phenylalanine hydroxylase (PAH) were published, the results of a long-term and successful collaboration between researchers at CSIC and CNIO in Spain, and the University of Bergen in Norway can finally be presented: full-length structures of this very important metabolic enzyme with and without its cofactor tetrahydrobiopterin (BH4). This work has been directed by Professors Juan A. Hermoso and Aurora Martinez with postdoctoral researchers Marte I. Flydal and Martin Alcorlo as shared first authors.
The PAH enzyme resides mainly in the liver where it removes excess of the amino acid phenylalanine from dietary proteins. Between meals, the enzyme is bound to BH4, a molecule that is essential for the enzymatic reaction to occur, but that also both stabilizes the complex structure of the PAH enzyme and lowers its activity when removal of the amino acid is not needed. Too much of the amino acid causes brain damage and too little of it hinders our own protein production and thus the BH4 molecule is one of the sophisticated ways in which the body regulates the activity of PAH.
In average, 1 in 10 000 newborns worldwide are born with a non-functional variant of the PAH enzyme and have the disease phenylketonuria, commonly known as PKU. They must live on a strictly controlled diet with limited amounts of protein their whole life to maintain a healthy brain. If the diet is not controlled, the increased fenylalanin levels in blood exerts toxic effects in the brain, manifesting as severe mental and psychomotor retardation, seizures, and psychiatric disorders. Even with a highly restrictive low protein diet to limit intake of the amino acid, children afflicted with PKU may experience more subtle neurodevelopmental and psychosocial problems resulting in a lower quality of life.
Most of the more than 950 PKU-associated mutations lead to a less stable and dysfunctional PAH enzyme that is more prone to be degraded. The stabilizing property of the BH4 molecule has been exploited and today oral administration of this molecule as a medicine called Kuvan® is the only readily available treatment option for PKU. Only about 25 percent of PKU patients are helped by Kuvan®, but they can tolerate a higher amount of dietary protein because the mutated PAH enzyme gains adequate stability to perform its function.
The PAH enzyme is composed of four identical subunits that must be quite flexible to allow the movements necessary for regulation of the enzymatic activity in response to phenylalanine levels. This flexibility deters the formation of the ordered crystals that must be made for structural determination and explains the previous unsuccessful attempts to crystallize the full-length enzyme. However, Flydal et al. also exploited the stabilizing property of BH4. By adding this molecule to the PAH enzyme they obtained crystals that in fact allowed them to solve two different structures at 3.18 Å resolution, one where BH4 is bound to all 4 subunits and one where BH4 is bound only to half of them.
The resulting structures are thus not only the very first full-length structures of the human enzyme, but also the first full-length structure of a PAH with BH4 bound to it. The PAH:BH4 complex is fundamental in normal enzyme functioning and in the treatment of PKU, and these structures will also serve to the development of even more effective therapies.
Original article
"Structure of full-length human phenylalanine hydroxylase in complex with tetrahydrobiopterin"
Authors: Marte Innselset Flydal, Martín Alcorlo-Pagés, Fredrik Gullaksen Johannessen, Siseth Martínez-Caballero, Lars Skjærven, Rafael Fernandez-Leiro, Aurora Martinez, and Juan A. Hermoso
PNAS, published 22 May 2019