New gene variants linked to congenital heart disease
Around 600 children are born with congenital heart disease in Norway each year. Little is known about the causes of congenital heart defects. A new international study involving researchers from University of Bergen and Haukeland University Hospital shows that the NAA15 protein can play an important role.

Main content
Congenital heart disease (CHD) is the most common type of birth defect, affecting about 1% of all newborns. This means that around 600 children are born with CHD in Norway each year. CHD reflects defects in the developmental program of the heart, which leads to malformations of the heart or the large blood vessels surrounding the heart. In most cases, no obvious cause of CHD is identified. Presumably, both heredity and environmental factors play a role. Defining the genetic causes of CHD will provide new insights into mechanisms of heart development that may eventually benefit patients with CHD and their families.
Defects in the NatA enzyme can lead to developmental defects
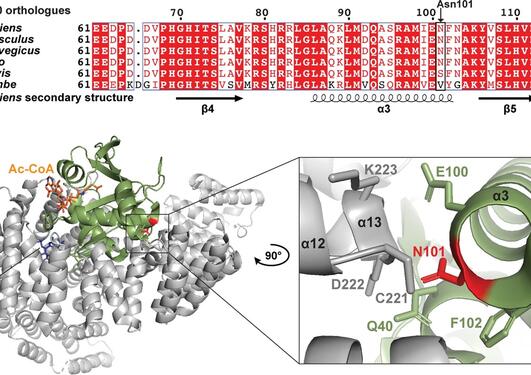
Genetic studies have shown that NAA15 variants may pose a risk of congenital heart defects. The NAA15 gene encodes the ribosomal subunit of an enzyme complex called NatA. NatA catalyzes the transfer of an acetyl group from acetyl-CoA to the start of a protein, a process called N-terminal acetylation. Acetylation the N-terminus is a prevalent modification and it is estimated that 85% of our proteins are to varying degrees acetylated at the N-terminus. The effect of N-terminal acetylation on protein is diverse, and includes changes to protein stability, complex formation, protein folding, and aggregation. Mutations that affect the NatA complex can lead to reduced N-terminal acetylation. This can affect heart and brain development, causing a wide range of congenital heart defects and neurological symptoms.
NAA15 a risk gene for congenital heart disease
In a new international study, published in the medical journal Circulation Research, researchers have investigated the relationship between N-terminal acetylation, NAA15 and CHD (Figure 1). By examining the genome of 4,511 CHD patients in the United States by whole exome sequencing, researchers at Harvard University identified four subjects with a rare loss-of-function variant in the NAA15 gene. Further investigations of these rare NAA15 variants in gnomAD, a population frequency database where exome and genome sequencing data from a variety of large-scale sequencing projects is collected, supported the suspicion that these variants increase the risk of CHD. The study, which was led by researchers from the Seidman Laboratory at Harvard Medical School, also identified 16 rare NAA15 missense variants (or mutations) that will alter the amino acid sequence in the gene product.
Yeast as a model system to study human disease
Yeast can be used as a model organism to study the function of genes involved in human disease. When yeast cells lack the NatA complex, they grow slower than normal cells and they become very sensitive to environmental changes, such as elevated temperatures. Molecular biologist Sylvia Varland has designed yeast cells that express the human NatA complex with different variants of NAA15.
- The growth of these designer yeast cells at elevated temperatures can serve as proxy for NatA activity, says Sylvia Varland a researcher at the Department of Biomedicine, University of Bergen.
The two NAA15 variants D335fs and S761* were not able to rescue the temperature-sensitive growth phenotype of yeast cells lacking the NatA complex (Figure 2). This finding suggests that the mutations cause loss-of-function. The NAA15 R276W variant, however, rescued yeast growth, pointing towards a partial NatA function in yeast.
Genetically engineered NAA15 stem cell model using CRISPR/Cas9
Our cells have two sets of genes, one from each parent. If a gene is deleted or inactivated, due to a loss-of-function mutation, the remaining functional gene variant may not be sufficient to produce the necessary gene product to ensure normal function. This phenomenon is known as haploinsufficiency (haplo = single, insufficiency = lack of).
Pluripotent stem cells are non-specialized cells that have the capacity to become any cell type in our body, such as heart muscle cells (cardiomyocytes). What happens if we affect the protein level of NAA15, will the stem cells still be able to differentiate into heart cells and will they function normally? In this study, researchers introduced NAA15 variants into induced pluripotent stem cells (iPS cells) using the gene editing tool CRISPR/Cas9. This resulted in iPS cells that were either lacking (NAA15-/-) or had reduced levels of NAA15 (NAA15+/- and NAA15+/ R276W). By adding a cocktail of various chemicals and growth factors, iPS cells can differentiate into heart muscle cells. The iPS cells that lacked NAA15 grew slowly and failed to differentiate into heart cells. iPS cells that only expressed one normal copy of the NAA15 gene differentiate into cardiomyocytes (NAA15+/- and NAA15+/R276W), but they did not have the same ability to contract as cells with normal levels of the NAA15 protein (NAA15+/ +).
- This study shows that the level of NAA15 and thus probably the level of NatA-mediated N-terminal acetylation of proteins is essential both for normal differentiation into cardiomyocytes and for these heart cells to have normal function, says Thomas Arnesen, who is Professor at the University of Bergen and Principal Investigator at the Department of Biomedicine and Department of Surgery, Haukeland University Hospital.
Ribosomal protein deficiency due to reduced level of NAA15
NAA15 is part of the enzyme complex NatA, which performs N-terminal acetylation during protein synthesis. The Gevaert lab at VIB-UGent performed proteomics analysis to assess the level of N-terminal acetylation in the NAA15 mutated iPS cells. Only a few NatA substrates were less acetylated in iPS cells not expressing NAA15. In contrast, there was a clear difference in the steady-state protein levels between normal cells and NAA15 mutated cells. In total, 562 proteins were differentially expressed in cells lacking or having reduced levels of NAA15 compared to normal cells.
- These findings suggest that NAA15 and the NatA complex play an important role in protein synthesis and/or protein stability, says Sylvia Varland.
NAA15 anchors the NatA complex to the ribosomes, which are molecular machines that perform protein synthesis. Deletion of NAA15 led to reduced levels of several ribosomal proteins, clustering near the NatA docking site. The binding of NAA15 to one or more of these proteins may protect them from degradation either during ribosome assembly or subsequent ribosomal activity. This suggests that deficiency or absence of NAA15 may lead to ribosomal protein deficiency, either by the lack of direct binding to ribosomal proteins or via reduced N-terminal acetylation of proteins. When the ribosomes do not function optimally it can lead to reduced protein production, which can affect normal cell development.
The Arnesen lab acknowledges support from the Research Council of Norway (RCN), the Western Norway Regional Health Authority, and the Norwegian Cancer Society. Sylvia Varland received a mobility grant from RCN which was cofunded by EU. Parts of the work was carried out at the Donnelly Centre for Cellular & Biomolecular Research, Toronto, Canada.
ORIGINAL PUBLICATION
Ward TL, Tai W, Morton SU, Impens F, VanDamme P, Van Haver D, Timmerman E, Venturini G, Zhang K, Jang MY, Willcox JA, Haghighi A, Gelb BD, Chung WK, Goldmuntz E, Porter GA, Lifton R, Brueckner M, Yost HJ, Bruneau BG, Gorham J, Kim Y, Pereira AC, Homsy J, Benson CC, DePalma SR, Varland S, Chen CS, Arnesen T, Gevaert K, Seidman CE, Seidman JG. Mechanisms of Congenital Heart Disease Caused by NAA15 Haploinsufficiency. Circulation Research 2021 in press, doi: 10.1161/CIRCRESAHA.120.316966.